|
Authors:
Dr. Mohammed Owaidha Al-Ajmi, MBBCh, MD*
Dr. Satheesh Kalanthra Kutty, MRCPCH, MD*
* Department of Pediatrics, Al-Jahra Hospital, Kuwait.
+ Department of Pediatrics

Key Words: EMA - Ethyl Malonic Aciduria.
MRI - Magnetic Resonance Imaging.
ABSTRACT
Ethyl malonic aciduria encephalopathy is a syndrome
characterised by relapsing petechiae and progressive neurodegenerative
symptoms and signs. The defining metabolic abnormality is
the excretion of large amounts of ethyl malonic acid in the
urine. EMA encephalopathy has been reported in fewer than
30 cases worldwide, the majority of them being in Europe,
Israel and Saudi Arabia. We report a patient with EMA presenting
with nephrotic syndrome and respiratory failure.
INTRODUCTION
Ethyl malonic encephalopathy (EE) first described
by Burlina ed al [1991] is a syndrome characterised by relapsing
petechiae, progressive neurodegenerative symptoms and signs,
acrocyanosis and in some cases with chronic diarrhoea. There
has been no case report with this syndrome presenting with
nephrotic syndrome and respiratory failure.
Marked persistent ethyl malonic and methylsuccinic
aciduria with abnormal excretion of C4-C5 (n-butyrl-; isobutyryl
-, isovaleryl -, and 2 methyl butyryl -) acylglycines and
acylcarnitines are typical biochemical findings. Excretion
of adipic acid is not increased in contrast to multiple acylcoenzyme-A
dehydrogenase deficiency. Ethyl malonic aciduria is also found
in short-chair acyl-coenzyme dehydrogenase deficiency. Symmetric
lesions in the basal ganglia (caudate and putamen), periventricular
white matter and cerebellar dentate nuclei are detected on
MRI (Ozano et al 1994; Burlina et al 1994).
The vascular manifestation of EE are unusual
and characteristic features. Acrocyanosis appears to be its
mildest manifestation. The development of showers of petechiae,
ecchymosis in response to intercurrent illness has lead to
investigation and treatment for presumptive meningococcaemia.
Biopsy of the skin lesion has shown only haemorrhage.
Further evidence of a bleeding abnormality is
microscopic haematuria and haemoperitoneum. Dilated tortuous
retinal vessels are seen.
Cerebral abnormality is manifest in infancy
as hypotonia and delayed development. Neurologic deterioration
accelerates following intercurrent infections, illness, and
most patients have died in the early years of life. Brain
imaging has shown infarcts of basal ganglia.
CLINICAL REPORT AND RESULTS
A 10 months old male infant was admitted
to the paediatric intensive care unit with acute respiratory
failure and generalised oedema. He was the sixth child of
healthy Kuwaiti parents who were first cousins. His four other
siblings were all healthy. He was born full term, appropriate
for gestational age by spontaneous vaginal delivery and had
a normal neonatal period. He appeared to develop normally
in the initial few months and at six months evaluation he
was noted to have neurodevelopmental delay. At this time he
was admitted to a regional hospital for evaluation, investigations,
and assessment.
On physical examination, growth was as follows:
Weight was 5th percentile, height 10th percentile and
head circumference 5th percentile. He had dysmorphic
features with depressed nasal bridge, epicanthal folds
and posteriorly rotated ears. A characteristic petechial
rash was evident on the face and limbs
(Fig.1). He had generalised oedema,
ascites and pleural effusion. Neurologically he had
a Glasgow coma scale of 7/15, generalised hypotonia
with increased deep tendon reflexes and bilateral sustained
ankle clonus.
|
|
INVESTIGATIONS
Haemoglobin 10 gms/L, haematocrit 0.306
units, bicarbonate 17 mmol/L, sodium, potassium and
chloride 131 mmol/L, 4.7 mmol/L and 110.8 mmol/L respectively,
a normal prothrombin time, partial thromboplastin time,
bleeding time and platelet count (248).
His serum lactate was high ( 7.0), his
plasma lactate to pyruvate-ratio was elevated (68.3)
(N <25), blood ammonia, and amino acids were normal.
His ANA, CANCA Ab, pANCA Ab and Antids-DNA (8A U/ml
(NR < 26, AU ml) were normal. Lysosomal enzymes,
very long chain fatty acids were normal.
Urine examination showed proteinuria +++,
and 24 hour urine protein was >10g/ 24 hours, spot
urine protein/creatinine ratio was raised (360 mgmmol),
S.albumin 17 g/L, S.Cholesterol was raised.
Urine for organic acids showed markedly
raised ethyl malonic acid. Blood spot acylcarnitine
showed an elevated concentration of butyrylcarnitine
(C4) and ratio of butylcarnitine to propionylcarnitine,
C4/C3 were substantially above normal.
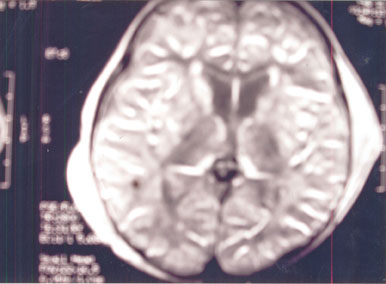
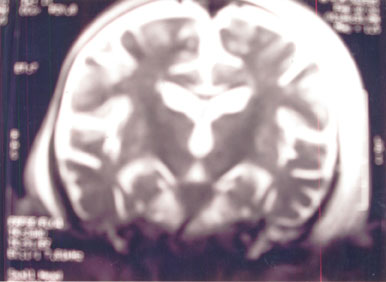
MRI brain revealed disseminated white
matter lesions, hypointense on T1W and hyperintense
on T2W & PD (Fig.2 & Fig.3).
The lesions involve cerebellum, medulla and cerebral
white matter. The caudate nuclei are also hyperintense
as T2 & PD.
EEG was abnormal with asymmetric background
and active focus from left frontotemporal leads.
Blood spot acylcarnitine and urine organic
acid analysis for parents and siblings were normal.
The analysis of the ETHE 1gene for the
patient revealed the presence of a homozygous deletion
of exon (4).
The parents and the healthy sibling showed
the presence of exon (4). It has been fully sequenced
and it was found to be homozygous normal, thus indicating
that the other allele in the index case was deleted.
ETHE I protein analysis was confirmed
by Western Blot analysis from skin fibroblast culture,
which revealed complete absence of ETHE 1 protein .
Based on the above clinical, biochemical,
MRI and urine findings a diagnosis of EMA with nephrotic
syndrome was made. Management included initial ventilatory
support from which he was successfully weaned off. The
nephrotic state was managed with steroids. The response
to steroids was noted over a period of four weeks. Dietary
regulation included methionine free milk formula9, carnitine
Vitamin E and ascorbic acid.
DISCUSSION
This 10 months old male infant is an unusual
case of ethyl malonic encephalopathy presenting with
nephrotic syndrome and in respiratory failure has evidenced
from the typical clinical, biochemical, MRI findings,
gene analysis, and urine examination. (1) Malgorzata
JM et al reported EMA rises from abnormal isoleucine
metabolism The observed high lactate/pyruvate ratio
in EMA suggested the possible involvement of the mitochondrial
electron transport chain (2) (Garavaglia et al 1994.
(3) Hoffman et al reported fatal progressive pancytopenia
with ethyl malonic acidurias in which were para crystalline
inclusion bodies on electron microscopy findings and
reduced activity of cytochrome C oxidase and reductase
in muscle. The blood counts in the index case were normal.
Progressive neurologic disease and partial deficiency
of cytochrome oxidase were reported by Lehnert and
(4) Ruitessbeck. (5)Burlina AB et al reported
this syndrome with normal fatty acid oxidation in fibroblast.
A relationship with this syndrome and the metabolism
of sulphur aminoacid has been proposed by (6)
Duran et al. We are reporting this case of EMA encephalopathy
with the unusual association with nephrotic syndrome
and has not been reported hitherto in the literature.
It is proposed that the vasculitis of EMA or EMA in
the urine can involve the glomerular basement membrane
causing protein loss. Electron microscopic study of
renal tissue may elucidate this process.
|
|
ACKNOWLEDGEMENTS
Dr. Fahad Al-Anizi
Pediatric Nephrologist
Al-Jahra Hospital.
Valleria Tiranti MD.
Division of Molecular Neurogenetics
National Neurological Institute "C. Betta"
Via Teniolo 4, 20126 Milano, Italy.
REFERENCES
- Malgorzata JM, Nowaczyk JM, Lehotay
DC et al. Ethyl malonic encephalopathy arises from
abnormal isoleucine metabolism. Metabolism. 1998;
35: 587 - 595
- Garavaglia B, Colamaria V. Carrara
F, et al. Muscle cytochrome c oxidase deficiency in
two Italian patients with ethylmalonic aciduria and
peculiar clinical phenotype. J Inherit Metab Dis.
1994; 17:301-303.
- Hoffman GF, Hunneman DH, Jakobs C,
et al. Progressive fatal pancytopenia, psychomotor
retardation and muscle carnitine deficiency in a child
with ethylmalonic academia. J. Inher Metab Dis. 1990;
13 : 337 - 340.
- Lehnert W, Ruitenbeek W. Ethylmalonic
aciduria associated with progressive neurological
disease and partial cytochrome c oxidase deficiency.
J Inher Metab Dis. 1993; 16:557 - 559.
- Burlina AB Dionisi = Visi C, Bennett
MJ, et al. A new syndrome with ethyl malonic aciduria
and normal fatty acid oxidation in fibroblasts. J.
Pediatr, 1994; 124 : 79 - 86.
- Duran M, DorlandL, van den Berg IET,
et al. The ethhylmalonic acid syndrome is associated
with deranged sulfur amino acid metabolism leading
to urinary excretion of thiosulfate and sulfothiocysteine.
In: Program and abstracts of the VII International
Congress of Inborn errors of metabolism; May 21 -
25, 1997; Vienna, Austria. Abstract 048.
- Nowaczyk MJM, Blaser SI, Clarke, JTR.
Central nervous system malformations in ethyl malonic
encephalopathy. AMJ Med. Genet. 1998; 75: 292 - 298.
- Ozano PT, Rashed M. Millington DS,
et al. Ethyl malonic aciduria : an organic academia
with CNS development and vasculopathy. Brain Dev,
1994 ; 16 : 12 - 22.
- Karen A. McGowan, MD; William L. Nyhan,
MD, PhD; Bruce A. Barshop, MD, PhHD and others. The
role of Methionine of Ethylmalonic Encephalopathy
with Petechiae. ARCH Neurol/vo9l 61, Apr. 2004.
|