|
Evaluation of the Child
with Short Stature
Abdulrazak
Abyad
Correspondence:
A. Abyad, MD, MPH, MBA, DBA, AGSF,
AFCHSE
CEO, Abyad Medical Center
Chairman, Middle-East Academy for Medicine
Email:
aabyad@cyberia.net.lb
Children with short stature are encountered
often in family practice. By definition, one
child in 33 has height measurements below the
third percentile for age. While this is often
defined as the lower limit of "normal,"
most of these children are, in fact, healthy
and growing adequately. Many will attain normal
stature as adults. The practitioner's task is
to identify the few children who are short as
a result of medical conditions that lead to
failure of normal growth.
Birth weight and length do not reliably predict
ultimate height and weight. Tanner and co-workers
(1) have reported a correlation co-efficient
of only 0.25 between birth length and ultimate
height. However, the correlation between height
and two years of age and ultimate height is
nearly 0.8.
Growth retardation may exist if
(1) Height is less than two standard deviations
from the mean for age.
(2) Growth velocity is less than two standard
deviations from the mean growth velocity for
age or,
(3) A pubertal growth spurt fails to occur within
two standard deviations of the usual time (2).
The causes of short stature are listed in (Table
1). An understanding of the typical pattern
of growth seen in each of these conditions is
helpful in evaluating the short child. Determining
the level of epiphyseal maturation is also useful.
The radiologic standards published by Gruelich
and Pyle are used most widely and are based
on the growth centres and epiphyses of the left
wrist and hand (3).
Table 1: Causes of Short Stature
Familial short stature
Constitutional growth delay
Chronic systemic disease
.......CNS
abnormalities
.......Congenital
heart disease
.......Respiratory
disease (asthma, cystic fibrosis)
Gastrointestinal disease (inflammatory bowel
disorder, celiac diseases)
Renal disease (renal tubular acidosis, chronic
renal failure)
.......Immune
deficiency
.......Chronic
anemia
Primordial growth delay
Chromosomal abnormalities
.......Down
syndrome
.......Turner
syndrome
Skeletal dysplasias
.......Osteochondrodystrophies
.......Pseudohypoparathyroidism
Environmental causes
.......Malnutrition
.......Psychosocial
deprivation
Endocrine disease
.......Hypothyroidism
.......Growth
hormone deficiency
.......Cushing
syndrome
|
Bone age is usually equal to chronological
age in familial and primordial short stature,
but is delayed in other causes of short stature.
Its major value is prognostic, as children with
delayed bone age have a better chance to attain
normal adult height than do short children whose
bone age is not delayed.
Bone age and height age are helpful in estimating
a child's growth potential. The younger the
bone age (the state of skeletal maturation),
the greater the remaining growth potential.
Bone age is determined by a radiologist, using
standard tables. Height age is obtained on a
growth chart by drawing a horizontal line from
the patient's height to the 50th percentile
line for height and then dropping a vertical
line to the baseline to measure the age (4).
Although a child may have a delayed height
age (HA) with respect to chronologic age (CA),
if the bone age (BA) is proportionately delayed
(CA > HA = BA), the ultimate height may be
equal to that of the child whose chronologic
age is equal to his height age and bone age
(CA = HA = BA). Comparison of chronologic age,
bone age and height age may be used to classify
causes of short stature (5).
Height prediction can be used to confirm suspicion
of abnormal growth. To predict a target adult
height, an adjusted midparental height is obtained
by averaging the parents' heights after first
adding 13 cm to the mother's height if the child
is a boy or subtracting 13 cm from the father's
height if the child is a girl. Projection of
the child's anticipated growth along his or
her growth percentile should yield an adult
height that is within + 8.5 cm of the adjusted
midparental height. If the projection of the
child's growth is more than 8.5 cm below the
adjusted midparental height, the growth of the
child cannot be assumed to be secondary to parental
short stature (6,7).
|
EVALUATION OF GROWTH RETARDATION |
History and Physical Examination
If growth retardation is suspected, particular
attention must be given to certain key aspects
of the history and physical examination. The
categories outlined in Tables 2 and 3 are touched
on in the well-child examination. However, in
the evaluation of growth retardation, each area
must be more extensively considered. For example,
a family tree can be used to plot family heights,
ages of menarche and ages of pubertal growth
spurts in search of familial short stature and
constitutional delay of growth and maturation.
Upper-to-lower segment ratios, usually not calculated
in general physical examinations, should be
included in all evaluations of growth in order
to detect abnormalities of bone development
(3).
Table 2: Important Historical Features in the
evaluation of the Child With Growth Retardation
| Historical
Features |
Diagnostic
Implications |
MATERNAL
HISTORY
Length of gestation, previous fetal abortions,
complications of TORCH infection, pregnancy,
smoking, alcohol and drug use |
Fetal
alcohol syndrome, hydantoin syndrome, intrauterine
growth retardation secondary
to placental insufficiency
|
ANTHROPOMETRIC
VALUES
Birth weight, birth length, dysmorphology
|
Intrauterine
growth retardation, Turner's syndrome, Down's
syndrome, other short stature syndromes |
NEONATAL
AND DEVELOPMENTAL HISTORY
Neonatal hypoglycemia, hypothyroidism
developmental milestones
|
Hypopituitarism,
|
NUTRITIONAL
HISTORY
Inadequate caloric intake |
Failure
to thrive
|
PSYCHOSOCIAL
HISTORY
Child neglect or psychological child abuse |
Environmental
|
FAMILY
HISTORY
Genetic syndromes. Skeletal dysplasias (e.g.
achondroplasia), inborn errors of metabolism
(e.g. mucopolysaccharidosis, gangliosidosis
type I, mucolipidosis II)
Family height, ages of menarche,constitutional
delay of maturation, ages of pubertal growth
spurts
|
Familial
short stature
|
REVIEW
OF SYSTEMS
Specific chronic organic diseases |
Cardiac,
pulmonary, hepatic,
disorders |
MEDICATION
HISTORY
Corticosteroids, stimulants |
Drug-induced
growth retardation
|
Table 3: Important Physical Findings in
the Evaluation of the Child with Growth Retardation
| Physical
Findings |
Diagnostic
Implications |
Upper-to-lower
segment ratio
|
Disorders
affecting bone growth (skeletal dysplasias)
or resulting in infantile proportions, (hypothyroidism,
hypopituitarism) |
Head
circumference
|
Evidence
of poor cerebral growth, (malnutrition) |
Goitre,
prolonged jaundice, large posterior fontanelle,
umbilical hernia
|
Hypothyroidism
|
| Micropenis,
visual disturbances |
Hypopituitarism |
| Heart
murmur, increased blood pressure, pallor,
wasting |
Evidence
of chronic organic disease
|
| Stigmata
of short stature syndromes |
|
Cubitus
valgus, webbed neck, low posterior hairline,
edema of hands and feet
|
Turner's
syndrome
|
Flat
facies, inner epicanthal folds, upward lateral
slant of palpebral fissures, short metacarpals
and phalanges, simian crease
|
Down's syndrome
|
Laboratory Evaluation
If growth retardation is suspected, the following
routine screening tests should be performed: Complete
blood count (anaemia); erythrocyte sedimentation
rate (inflammatory bowel disease, which may be
relatively asymptomatic except for growth retardation);
urinalysis, blood urea nitrogen, serum creatinine
and serum bicarbonate (renal disease); thyroid
function tests (hypothyroidism) and hand films
for bone age (helpful in determining whether growth
is consistent with chronologic age and in excluding
skeletal dysplasias) (9).
If a child with growth failure is more than
two years old, the plasma somatomedin-C (SM-C)
level can be determined to screen for growth
hormone deficiency. An SM-C level of less than
0.25 u per
mL suggests a growth hormone deficiency. A value
greater than 0.5 u
per mL indicates that a growth hormone deficiency
is unlikely. However, the SM-C level is not
useful during the first two years of life because
of the overlap of levels between normal and
growth hormone-deficient patients (10).
If the SM-C level is abnormal or if there
is strong suspicion of a hormonal deficiency
(hypoglycemia in a short child), growth hormone
stimulation tests should be done. Since baseline
growth hormone levels are low, stimulation tests
are required to separate subjects with hormonal
deficiency from those with normal secretion
(11).
Metabolic screening tests should be performed
as needed to identify mucopolysaccharidosis,
aminoacidopathies and galactosemia. Any girl
with delayed bone age and unexplained shortness
should have a karyotype done to rule out Turner's
syndrome or one of its variants.
Table 4: Laboratory Evaluation
| Initial
Evaluation |
Complete
blood count
Serum chemistry
Urine analysis
Wrist X-ray for bone age |
| Further
Tests as Necessary |
Chromosome
analysis
Lateral skull X-ray
Thyroxin and thyroid
Stimulating hormone
Growth hormone level stimulation tests |
Differential Diagnosis
A useful way of approaching growth disturbances
is by comparing chronologic age, bone age and
height age (Table 5). A bone age that differs
from height age by six months or less is not
significant. However, a bone age that differs
from height age by one year or more is significant
(12).
Table 5: Differential Diagnosis of Growth
Disorders by Comparing Chronologic Age, Bone
Age, and Height Age
| CA >
BA = HA
Hypopituitarism
Constitutional delay of growth and maturation
Nongrowth-hormone-deficient, growth-hormone-responsive
growth failure (biologically inactive
growth hormone, or growth hormone and/or
somatomedin-C resistance)
Cushing's disease
Chronic malnutrition
Psychosocial deprivation
Chronic organic disease
Glucocorticoid excess
CA > HA >
BA
Growth hormone
deficiency (hypopituitarism)
Hypothyroidism
CA > BA >
HA
Constitutional
delay of growth and maturation with familial
short stature
Intrauterine growth retardation
Turner's syndrome
Down's syndrome
CA = BA >
HA
Familial
short stature
Intrauterine growth retardation
|
KEY: CA = chronologic age; BA = bone age;
HA = height age
Treatment of short stature depends on the underlying
cause. Children with chronic systemic disease
will show improved growth if their medical status
can be significantly improved. Growth failure
because of dietary or environmental factors
can also be reversed with appropriate intervention.
Children with hypothyroidism usually show a
rapid return to normal stature once hormone
replacement is begun. For children with growth
hormone deficiency, however, the results of
treatment are seldom as dramatic, with most
individuals remaining subnormal in height as
adults.
There is no specific treatment for the other
causes of short stature. Nevertheless, parents
may enquire about the benefit of growth hormone
treatment. When the child has constitutional
delay, parents can be reassured that the adult
height will be normal without intervention.
Unfortunately, the same cannot be said for those
with familial or primordial short stature. There
is no evidence to show that the use of growth
hormone results in any significant increase
in final height for these children.
Growth hormone does seem to offer possible
benefit to children with Turner syndrome. Although
the results of long-term studies are not yet
available, most girls with this condition who
have been given growth hormone have shown an
increase in linear growth that is expected to
result in significant improvement in their adult
height (13). Referral to a pediatric endocrinologist
seems appropriate when Turner syndrome is diagnosed.
In the second half of this paper I will present
a charming child with short stature. The workup
of this patient demonstrates the step that should
be followed in investigation of short stature.
| HISTORY
AND PHYSICAL EXAMINATION |
Haifa was seen initially in Tripoli and was
referred later to AUB where I saw the patient
in the Family Medicine Practice Center. The
investigations were done in Tripoli, AUB and
Royal Hospital for sick children in London.
She was born in Tripoli in a maternity hospital,
birth weight 3 kg, birth length 49 cm, following
a spontaneous vertex delivery. Mother had been
well during the pregnancy with no smoking nor
alcohol intake. Mother is aged 36 with a height
of 163.0 cm which is 50th centile. She reached
menarche at age 13 years. Father, aged 37 years,
is an agricultural land owner in good health.
He is 183 cm tall, which would put him between
the 9th and 97th centiles. There are four siblings,
a boy aged 16 years who is 183 cm tall, a girl
of nearly 13 years who has been menstruating
for some six months, she is 167 cm tall, a girl
of 10 years, said to be 154 cm, and a boy of
10 months, said to weigh 12 kg and be around
74 cm tall some two months ago. There is no
history of stillbirth, neonatal death nor death
in infancy.
In the past, Haifa has been in good health.
She was breast fed for six months and solids
introduced by seven to eight months of age.
However, mother says that her appetite has always
been bad and that she often has to force food
into her. Bowels are open regularly once a day,
said to have been of rather small volume but
more normal of the last year and normal in colour.
There have been no serious illnesses. Mother
had no height records but thought that she had
gained 5 cm over the last year and 8 cm the
year before. She thinks her weight has been
static over the last two years.
On examination, Haifa was a delightful girl
who looked well. She looked somewhat dysmorphic
with coarse features, hypertelorism, rather
square face with snub nose and coarse hair.
Limbs superficially looked short with particularly
short fingers and square hands with broad great
toes. No abnormalities were found in the central
nervous system with normal fundi, no cataracts,
no squint. The cardiovascular system: There
was a systolic murmur heard over the precordium
at the left sternal edge and at the back with
no thrill. There was no femororadial delay.
There were no abnormalities in the respiratory
system nor the abdomen. In particular, there
was no hepatosplenomegaly. Genitalia were those
of the normal female and there was no kyphoscoliosis.
Triceps skinfold thickness was 6.2 mm (3rd centile),
subscapular skinfolds 4.2 mm (3rd to 10th centile).
Height 95.9 cm, which is well below the 3rd
centile, sitting height 56.8 cm, and subischial
leg length 39.1 cm, indicating that her back
and limbs are proportionately small (-3.5 and
-4 standard deviations, respectively).
Investigations
Haemoglobin, full blood count, showed no abnormality,
with ESR of 10 mm/hr. Serum iron level was 12
mol/l (nl) with normal transferrin (2.6 g/l)
and ferritin level (52 ug/l). Both vitamin B12
and folate level were within normal 824 ng/l
and 10.6 ug/l, respectively with red cell folate
of 371 ug/l (normal). Her electrolytes profile
was normal including: Calcium 2.42 mmol/l, phosphate
1.38 mmol/l, creatinine 5.3 mmol/l. The plasma
amino acid screen and organic acid urinary screen
were normal as well. Qualitative urinalysis
revealed trace of protein and ketones but no
other abnormality. Mucopolysaccharidosis screen
revealed mucoolysaccharides of 19 mg/mmol creatinine
(age related reference range 6-13 mg/mmol creatinine)
The pattern obtained from one dimensional
electrophoresis does not support the diagnosis
of mucopolysaccharidosis types I, II, or III.
Her thyroxin was 165 nmol/l (normal) and prolactin
307 m u/l (normal).
Combined GnRH and TRH stimulation tests:
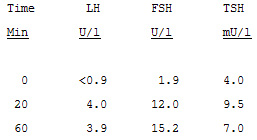
These are normal results though there is a somewhat
exaggerated response of FSH of doubtful clinical
significance.
Insulin hypoglycemia test: Plasma glucose
fell from 4.7 mmol/l to a minimum of 1.8 mmol/l.
Cortisol response baseline 409 nmol/l, maximum
814 nmol/l (normal response). Growth hormone
rose to maximum of 16.5 mU/l (very slightly
suboptimal response, but unlikely to be of clinical
significance).
Jejunal biopsy: Two attempts failed,
but stool culture negative. In addition, urine
for reducing substances was negative.
Skeletal survey: There is abnormality
of the hands and feet. This consists in the
hands of short metacarpals, phalanges and a
small carpal area. Similar changes are present
in the feet. Long bones show only minor abnormality
with slight loss of tubulation of the proximal
humeri and tibiae. Remaining skeleton including
the skull and spine, normal. Appearances are
not of a mucopolysaccharidosis, but suggest
a possible acrodysplasia.
It seemed very likely, on the first meeting
with Haifa, that she had a syndrome diagnosis
and I felt it quite possible that she would
turn out to have either a mucopolysaccharidosis
or a form of skeletal dysplasia. The investigations,
however, have excluded a mucopolysaccharidosis
and the measurements indicate that her short
stature is proportional, with back and long
bones equally short. The skeletal survey findings
are not pathognomic of any particular syndrome
and I would have expected the radiological changes
to be more specific by her age in conditions
such as acromesomelic dysplasia syndrome or
brachydactyly syndrome type-E, although the
latter does remain a possibility (14, 15).
The importance of making a specific syndrome
diagnosis would be to provide genetic counselling
and, of course, to be more specific about the
prognosis. Most important, however, is the question
as to whether any treatment is likely to influence
Haifa's final height and, despite the marginally
suboptimal growth hormone response to insulin
hypoglycemia, there is nothing clinically about
Haifa to suggest that she might respond to exogenous
growth hormone treatment. There is nothing from
our other investigations to suggest any other
form of treatment that is likely to prove beneficial.
In summary, therefore, I think that Haifa
is small because of a dysmorphic syndrome which
we have been unable to positively identify.
I think that it is very unlikely that any therapeutic
intervention will improve her final height prognosis.
Final Comment
Growth is a manifestation of health in the young.
As such, it is a parameter of the well-being
of a child. A wide variety of disorders can
affect the rate and the quality of growth. Thus,
the ability to evaluate growth is a basic diagnostic
skill that all physicians who provide care for
children should possess. By focusing on key
aspects of the history and physical examination,
by performing the appropriate screening tests,
by comparing chronologic age, bone age and height
age, and by reviewing prior anthropometric measurements,
the family physician can confidently evaluate
the child with growth retardation.
1.
Tanner
JM,
Healy
MJ,
Lockhard
RD,
MacKenzie
JD,
Whitehouse
RH.
Alberdeen
growth
study
I.
The
prediction
of
adult
body
measurement
from
measurements
taken
each
year
from
birth
to
five
years.
Arch
Dis
Child
1956;31:372-81.
2.
Frasier
SD.
Growth
disorders
in
children.
Pediatr
Clin
North
Am
1979;26:1-14.
3.
Greulich
WW,
Pyle
SI.
Radiographic
atlas
of
skeletal
development
of
the
hand
and
wrist,
2nd
ed.
Stanford
CA:
Stanford
University
Press,
1959.
4.
Lindgren
G.
Growth
of
schoolchildren
with
early,
average
and
lage
ages
of
peak
height
velocity.
Ann
Hum
Biol,
1978;5:253-67.
5.
Moore
DC.
Proportionate
delayed
growth.
In:
Kelly
VC,
ed.
Practice
of
pediatrics,
Vol
7,
chap
62.
Philadelphia:
Harper
&
Row,
1986:1-12.
6.
Himes
JH,
Roche
AF,
Thissen
D,
Moore
WM.
Parent-specific
adjustments
for
evaluation
of
recumbent
length
and
stature
of
children.
Pediatrics
1985;75:304-13.
7.
Roche
AF,
Wainer
H,
Thissen
D.
The
RWT
method
for
the
prediction
of
adult
stature.
Pediatrics
1975;56:1026-33.
8.
Linder
B,
Cassorla
F.
Short
stature:
Etiology,
diagnosis,
and
treatment.
JAMA
1988;260:3171-5.
9.
Mahoney
CP.
Evaluating
the
child
with
short
stature.
Pediatar
Clin
North
Am
1987;34:825-49.
10.
Kaplowitz
PB,
D'Ercole
AJ,
VAn
Wyk
JJ,
Underwood
LE.
Plasma
somatomedin-C
during
the
first
year
of
life.
J
Pediatr
1982;100:932-4.
11.
Moore
DC,
Rubalcaba
RH,
Smith
EK,
Kelley
VC.
Plasma
somatomedin-C
as
a
screening
test
for
growth
hormone
deficiency
in
children
and
adolescent.
Horm
Res
1982;16:49-55.
12.
Lipsky
MS,
Horner
JM.
The
child
with
short
stature.
Am
Fam
Physician
1988;37:230-41.
13.
Lippe
B,
Frasier
SD.
How
should
we
test
for
growth
hormone
deficiency,
and
whom
should
we
treat?
[Editorial]
J
Pediatr
1989;115:585-7.
14.
Vincent
M,
Riccardi;
MD,
Lewis
R.
Holmes,
MD.
Brachydactyly,
Type
E:
Hereditary
shortening
of
digits,
metacarpals,
metatarsals,
and
long
bones.
J
of
Pediatrics
1974;84:251-4.
|

